Dwa główne typy wielotorbielowatości nerek są klasyfikowane według ich
wzorów dziedziczenia (bardzo podobne do siebie objawowo): wyróżnia się
wielotorbielowatość nerek dziedziczoną autosomalnie dominująco (ang. autosomal dominant polycystic kidney disease, ADPKD) i postać autosomalną recesywną (autosomal recessive polycystic kidney disease,
ARPKD). Choroba ta występuje u ludzi i innych zwierząt.
Wielotorbielowatość nerek objawia się obecnością licznych torbieli w obu
nerkach. Choroba zajmuje również wątrobę, trzustkę, rzadko serce i mózg. Etiopatogenezę stanowią mutacje przynajmniej w 2 genach: PKD1 i PKD2, kodujących, odpowiednio, białka policystynę-1 i policystynę-2. Gen PKD1 znajduje się w locus 16p13.3, locus genu PKD2 to 4q13-23. ARPKD jest spowodowana mutacją genu PKHD1 kodującego fibrocystynę. Wiek rozpoznania wielotorbielowatości, ciężkość jej przebiegu i obecność powikłań pozanerkowych zależy od rodzaju mutacji.
W postaci PKD dziedziczonej autosomalnie dominująco w obrazie
klinicznym oprucz zwyrodnień nerek obserwuje się objawy, takie jak: torbiele wątroby, najczęściej bezobjawowe, torbiele śledziony, trzustki i płuc (rzadziej), tętniaki tętnic podstawy mózgu, wypadanie płatka zastawki mitralnej
lub inne zastawkowe wady serca (niedomykalność zastawki mitralnej,
niedomykalność zastawki trójdzielnej), tętniaki aorty, uchyłkowatość
jelita grubego, przepukliny brzuszne.
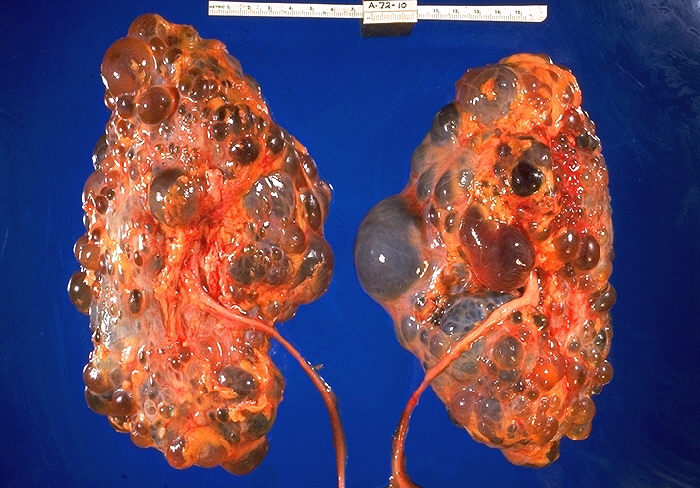 |
| Nerki chorego na wielotorbielowatość nerek w preparacie sekcyjnym |
Brak komentarzy:
Prześlij komentarz